
Study design, patient eligibility, treatment and oversight
Table of Contents
A phase 1, multicenter, open-label, first-in-human trial of ELI-002 2P monotherapy was conducted in five ascending dose cohorts at seven centers in the United States between 4 October 2021 and 24 September 2024 (the clinical cutoff date for the results presented here). A fixed dose of Amph-Peptides 2P (G12D and G12R, 0.7 mg each) was administered with escalating doses of 0.1, 0.5, 2.5, 5.0 and 10.0 mg Amph-CpG-7909 adjuvant. Eligible patients were 18 years or older, had mKRAS G12D-mutated or G12R-mutated pancreatic or colorectal cancers and were at high risk for relapse because of the presence of MRD (indicated by ctDNA-positivity or elevated serum CA19-9 and/or CEA). Clinical data was entered into Medidata Rave 2018.2.4. Additional details are provided in the study protocol (Supplementary Data 1).
At two institutions, City of Hope and the University of Colorado School of Medicine, central institutional review board (IRB) approval was obtained from WIRB Copernicus IRB. Local IRB approvals were provided for Memorial Sloan Kettering Cancer Center’s IRB, the University of Texas MD Anderson (Office of Human Subject Protection), the University of Iowa (Human Subjects Office/IRB), Northwell Health (Feinstein Institutes for Medical Research, Northwell Health IRB), the University of California, Los Angeles (Office of the Human Research Protection Program) and Massachusetts General Hospital (Dana–Farber Cancer Institute Office for Human Research Studies). The US Food and Drug Administration approved the study, which was registered on ClinicalTrials.gov (NCT04853017).
Patients
We enrolled adult (≥18 years old) patients with Eastern Cooperative Oncology Group performance status of 0 or 1 with pathologically confirmed mKRAS (G12D or G12R) PDAC or CRC, who were MRD+ with either (1) absolute CA19-9 ≥ 90 U ml−1 or cea ≥ 15 of ml−1 or (2) successively rising values (≥1 week apart) in either CA19-9 or CEA not attributable to a noncancer condition, such as pancreatitis, peritonitis, postoperative leak/fistula or biliary obstruction. Patients had recovered from prior surgery, chemotherapy or radiation without ongoing medical/surgical issues and were willing to use effective methods to avoid pregnancy and provided written informed consent. Baseline absolute neutrophil count ≥1.5 × 109 l−1platelets ≥100 × 109 l−1normal range liver function tests, serum creatinine <1.5 (or if serum creatinine was ≥1.5 mg dl−1creatinine clearance calculated by the Cockcroft–Gault formula ≥60 ml min−1 was acceptable), albumin ≥2.5 g dl−1 and IL6 <500 pg ml−1 were required.
PDAC patients had high risk tumor stages I, II, III or stage IV oligometastatic disease per current American Joint Committee on Cancer criteria with no evidence of disease on current imaging (equivocal radiographic findings such as subcentimeter lesions or potential resolving soft tissue changes after surgery were accepted), prior treatment with standard chemotherapy/chemoradiation administered in the neoadjuvant and/or adjuvant setting, and complete tumor resection (R0 or R1 pathologic margins), with focal use of intraoperative irreversible electroporation permitted.
CRC patients had high risk stage II (T4N0), stage III (T4N1-2/TanyN2) or stage IV oligometastatic disease per current American Joint Committee on Cancer staging criteria, prior cytotoxic chemotherapy administered in the neoadjuvant or adjuvant setting, or as total neoadjuvant therapy, and complete surgical resection (R0 or R1 pathologic margins), with focal use of intraoperative irreversible electroporation permitted.
We excluded patients who received antitumor therapy within 4 weeks, who had history of brain metastasis, other malignancies within the last 3 years (except for adequately treated carcinoma of the cervix, bladder, prostate, basal or squamous cell skin cancer), were receiving immunosuppressive drugs, those with serious comorbid illness including uncontrolled infection, class III or IV (New York Heart Association) cardiac failure, myocardial infarction within 6 months, active seizure disorders, autoimmune diseases or interstitial lung disease if requiring systemic steroids, pulse oximetry <92% on room air, prior organ transplants, HIV/AIDS, hepatitis B, hepatitis C (unless they had a sustained virologic response to direct-acting antiviral therapy) and those in the first two weeks of SARS-CoV-2. Women were excluded if pregnant or lactating. PDAC patients were excluded when tumors were of neuroendocrine subtype, or when there was a germline BRCA 1/2 mutation; CRC patients were excluded when tumors were mismatch repair defective (MSI+).
Treatment was divided into a ‘prime immunization series’ (six subcutaneous doses of ELI-002 2P over 8 weeks), a 3-month ‘no dosing period’ (observation) and a ‘booster immunization series’ (4 weekly doses of ELI-002 2P). A follow-up period of up to 2 years was included after the first dose of ELI-002 2P to monitor safety and efficacy.
The study was sponsored and designed by Elicio Therapeutics in collaboration with the academic authors. The study and analyses were conducted in accordance with the general principles of the Declaration of Helsinki and Good Clinical Practice guidelines of the International Council for Harmonization. The trial protocol, amendments and supporting documents were approved by the local/central institutional review board for each study site, the US Food and Drug Administration and were registered on Clinicaltrials.gov (NCT04853017). All patients provided written informed consent.
A safety and monitoring committee was convened to review safety and determine dose escalation and cohort expansion decisions. Cohorts ranged from three to six patients with expansions allowed after the first three patients completed 28 days without dose-limiting toxicity and when additional eligible patients had been identified.
All the authors affirm that the trial was conducted in accordance with the study protocol and vouch for the accuracy and completeness of the data. All the authors reviewed and revised the manuscript and made the decision to submit it for publication.
The initial protocol (version 1.0) was approved on 13 July 2020. Key protocol amendments are as follows: Amendment 2 (version 3.0) was approved on 23 February 2021 and included changes requested by the US Food and Drug Administration. This was the initial protocol for initiating the study. On 8 April 2022, Amendment 4 (version 5.0) was approved and added serum tumor biomarkers (that is, CEA and CA19-9) to the MRD eligibility along with ctDNA. Amendment 5 (version 6.0), approved on 2 August 2022, added language regarding pseudo-progression and continued ELI-002 dosing. Amendment 6 (version 7.0) was approved on 25 January 2023 and added language for public record search for OS. Amendment 7 (version 8.0) was approved on 7 August 2023 and added another year of follow-up to collect additional RFS and OS.
Endpoints and assessments
Primary endpoints of the study were safety (adverse events were graded per Common Terminology Criteria for Adverse Events, version 5.0), tolerability and determination of the RP2D. Secondary and exploratory endpoints include tumor biomarker reduction and clearance defined through assessment of ctDNA and/or serum tumor antigens (CA19-9 or CEA), radiographic relapse-free survival, defined as the time from initiation of ELI-002 treatment until confirmed radiographic progression using iRECIST criteria, and OS, and immunogenicity.
Immunogenicity analysis
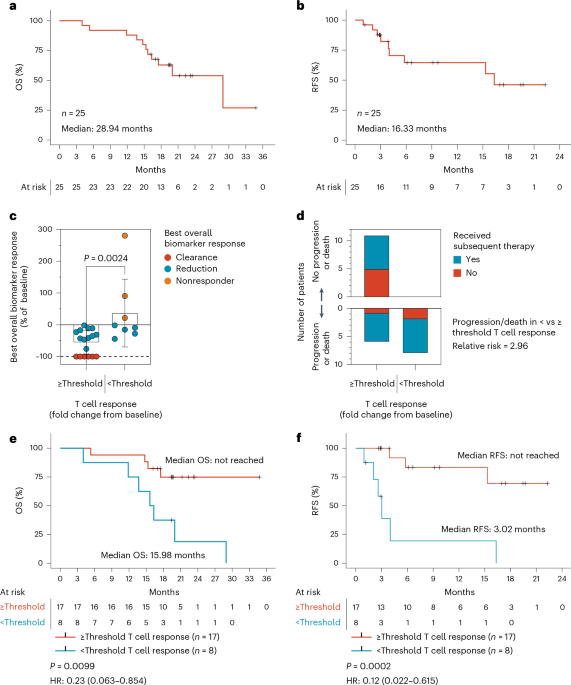
PBMCs for immunogenicity analysis were processed from leukapheresis (baseline, week 9) or whole-blood collections (all other timepoints). Patient PBMCs were processed by the Ficoll-Hypaque gradient protocol for leukapheresis samples or cell processing tubes (BD) for whole-blood samples. PBMCs were resuspended in CS10 freezing media (Cryostor), frozen in aliquots of 10–20 million cells per cryovial and stored in a temperature-monitored liquid nitrogen vapor phase freezer. Only PBMCs collected before subsequent therapy are included in datasets and graphs, with the exception of the long-term duration graphs (Fig. 2g,h). The maximum T cell response was determined as the maximum fold change from baseline to any postvaccination timepoint in either the ‘Ex vivo FluoroSpot assay’ or ‘Ex vivo ICS assay’ for any of the seven mKRAS antigens or a pool of all seven antigens combined. Figure 2A includes data from all T cell responders (n = 21/25). Figure 2B contains data from all responders with FluoroSpot data (n = 19/25) while Fig. 2c contains data from all responders with ICS data (n = 17/25). Figure 2g contains data from all patients with booster doses that had responses in FluoroSpot assay (n = 8) while Fig. 2h contains data from the boosted patients in Fig. 2g that were also tested for memory markers in the intracellular cytokine staining (ICS) assay (n = 6/8).
Ex vivo FluoroSpot assay
A direct IFNγ/granzyme B (GrB) FluoroSpot assay was performed on thawed PBMCs. Cryopreserved PBMCs were thawed in 10% human AB serum/RPMI media + Benzonase and rested overnight at 37 °C. Precoated human IFNγ/GrB FluoroSpot plates were washed with phosphate-buffered saline and blocked with AIM-V media for at least 30 min (MabTech). The 2 × 105 rested PBMCs were plated into each well and stimulated for 44 h as per the manufacturer’s instructions with seven individual mKRAS peptide pools and a WT peptide pool. Each pool consisted of a KRAS 18-mer peptide along with the corresponding 9-mer and 10-mer overlapping peptides (OLPs), at a concentration of 2 µg per peptide per ml. No exogenous cytokines were added to the PBMCs during this assay. All samples were plated in triplicate. Dimethyl sulfoxide was used as the negative control (background wells) and anti-CD3 (MabTech) was used as the positive control. The plate was developed based on the manufacturer’s instructions. Plates were scanned and counted using the IRIS plate reader (MabTech) using the FITC and Cy3 filters. Data are background subtracted, averaged per triplicate measurements and normalized to 1 × 106 PBMCs. A postvaccination sample was characterized as positive if it was at least 2 s.d. above the DMSO negative control. A responder in the FluoroSpot assay was defined as a patient with a ≥2-fold increase from baseline at any postvaccination timepoint and more than the minimum threshold of 50 total IFNγ and GrB spot-forming cells per 1 × 106 PBMCs.
Ex vivo ICS assay
A direct ICS assay for IL2, IFNγ and TNF was performed by flow cytometry. PBMCs were thawed and rested overnight. In total, 106 PBMCs per well were plated and stimulated for 17 h at 37 °C with individual mKRAS peptide pools at 2 μg ml−1 per peptide (Supplementary Table 1). GolgiStop and GolgiPlug (BD) were also added to each well. The next day, cells were surface stained with antibodies against CD4 (BV421—clone, SK3; BD, 566907; 2.5 μl per well), CD8 (BV786—clone, RPA-T8; BD, 563823; 1:25), CD45RA (Alexa 700—clone, HI100; BioLegend, 304120; 1:25), CCR7 (PE-CF594—clone, 15053; BD, 562381; 1:12.5), Aqua Live/Dead marker (Thermo Fisher Scientific, L34966; 0.5 μl per well) and dump markers CD14 (PE-Cy5—clone, 61D3; Thermo Fisher Scientific, 15-0149-42; 1:200), CD16 (PE-Cy5—clone, 3G8; BioLegend, 302010; 1:200), and CD19 (PE-Cy5—clone, SJ25C1; BioLegend, 363042; 1:100). Cells were subsequently fixed with CytoFix/CytoPerm (BD) and further stained with antibodies against CD3 (APC-H7—clone, SK7; BD, 560176; 2.5 μl per well), IFNγ (FITC—clone, Mab11; BioLegend, 506504; 1:200), TNF (BV711—clone, B27; BioLegend, 502940; 1:50) and IL2 (BV650—clone, MQ1-17H12; BioLegend, 500334; 1:50). Cells fixed in 0.5% formaldehyde were acquired on a BD FACSymphony and data were analyzed with BD FlowJo V10 software (gating progression and example plots in Supplementary Fig. 1). A responder in the ICS assay was defined as a patient having ≥2-fold increase in total IFNγ, IL2 and TNF from baseline at any postvaccination timepoint, along with a cytokine+ T cell frequency of ≥0.1%.
Some patients were also tested in an extended ‘Ex vivo ICS assay’ that included additional activation and cytotoxic markers. The extended ‘Ex vivo ICS assay’ was set up using the same methods as above, with the addition of CD107a (Alexa Fluor 700—clone, H4A3; BD, 561340; 1.25 μl per well) during the 17 h stimulation. The next day, cells were surface stained with antibodies against CD8 (BUV805—clone, SK1, BD, 612889), CD45RA (PE-Cy7—clone, HI100; BD, 560675), CCR7 (BUV615—clone, 3D12; BD, 562381), Aqua Live/Dead marker (Thermo Fisher Scientific, L34966; 0.5 μl per well) and dump markers CD14 (PE-Cy5—clone, 61D3; Thermo Fisher Scientific, 15-0149-42; 1:400), CD16 (PE-Cy5—clone, 3G8; BioLegend, 302010; 1:100) and CD19 (PE-Cy5—clone, SJ25C1; BioLegend, 363042; 1:100). Cells were subsequently fixed with the FoxP3/transcription factor staining buffer set (Thermo Fisher Scientific) and further stained with antibodies against CD3 (APC-H7—clone, SK7; BD, 560176; 1:40), CD4 (BUV496—clone, SK3; BD, 612936; 1:40), IFNγ (BB700—clone, B27; BD, 566394; 1:80), TNF (BV750—clone, MAb11; BioLegend, 502940; 1:80), IL2 (BV421—clone, MQ1-17H12; BD, 564164; 1:40), granzyme B (FITC—clone, GB11; BD, 560211; 1:40), perforin (PE—clone, B-D48; BioLegend, 353304; 1;80), CD137 (BUV661—clone, 4B4-1; BD, 741642; 1;80), CD154 (BUV563—clone, TRAP-1; BD, 748984; 1:80), CD69 (BV711—clone, FN50; BD, 563836; 1:160), Ki67 (BV650—clone, B56; BD, 563757; 1;80) and FoxP3 (PE/Dazzle 594—clone, 206D; BioLegend, 320126; 1:160).
Tumor biomarker assessment and mutation identification
Comprehensive genomic profiling, whole-exome sequencing (WES), was performed to determine whether the patient’s tumor harbored at least one of the two mKRAS alleles targeted by the ELI-002 2P (G12D or G12R). The Natera Signatera ctDNA test evaluated for the presence or absence of circulating tumor DNA. WES was performed on formalin-fixed, paraffin-embedded tumor samples with at least 20% tumor content confirmed by a pathologist under the Central Lab Improvement Amendments and College of American Pathologists guidelines. Genomic DNA was extracted from the patient’s normal (whole blood) and tumor tissue. Libraries of tumor and matched germline DNA were prepared and exomic regions were captured. The assay was performed by target enrichment of the isolated DNA, followed by 440× coverage sequencing on an Illumina HiSeq 2500 or NovaSeq 6000 (Illumina). Somatic single-nucleotide variants (SNVs) that were present in the tumor and absent in the germline were identified. A proprietary Natera algorithm selected a set of 16 SNVs to maximize the detectability of tumor DNA if present in plasma. Polymerase chain reaction primers targeting the 16 personalized SNVs were designed and synthesized to be used to identify and track ctDNA in a patient’s plasma. Cell-free DNA was extracted from plasma and analyzed using a multiplexed personalized polymerase chain reaction assay. Plasma samples with ≥2 SNVs detected above a predefined confidence threshold were deemed ctDNA+and ctDNA concentration was reported as mean tumor molecules per milliliter of plasma. In patients without adequate tumor tissue, a plasma-based ctDNA assay for mKRAS variants was performed using Sysmex SafeSEQ RAS-RAF. Cell-free DNA was isolated from plasma and a next-generation sequencing-based assay that evaluated K/NRAS to detect SNVs was performed using NextSeq 550 (Illumina). The ctDNA concentration was reported as the number of mutant molecules per variant and the mutant allele frequency. Local testing was permitted if already available to confirm mKRAS status. Serum tumor biomarkers, CA19-9 and CEA, were analyzed by the local laboratories at each study site.
Antigen-spreading assay
To assess for antigen spreading, PBMCs were stimulated in the ‘Ex vivo FluoroSpot assay’ and ‘Ex vivo ICS assay’ as above, with neoantigens not included in the ELI-002 2P vaccine. Genomic DNA is extracted from the patient’s normal and tumor samples using next-generation sequencing WES. Using the WES data for each patient, somatic single-nucleotide mutations present in the tumor and absent in the germline genomic DNA were identified using a validated bioinformatics tool (GEM ExTra pipeline NG2-LDT 1.14.0; Natera). The reference genome assembly used for alignment is NCBI GRCh37. Stop-gain and start-loss mutations were excluded. Up to ten neoantigens were randomly selected from the list of somatic SNVs generated by WES for each tested patient for antigen spreading testing. An algorithm was not used to select these neoantigens. First, an 18-mer was designed (generally with the mutation centered in the middle) and then Genscript synthesized two 15-mers overlapping by 11 to cover the mutated 18-mer (18-mer sequences found in Supplementary Table 1).
Statistical analysis
Descriptive statistics were used to summarize demographic, medical history and safety data. Continuous variables were summarized using mean, s.d., median, minimum value and maximum value. Categorical variables were summarized using frequency counts and percentages. Clinical efficacy outcomes, such as tumor biomarker reduction or clearance, were examined for association with categorical variables, including high versus low T cell response, using the Mann–Whitney test. The Kaplan–Meier method was used to estimate the survival distributions. The log-rank test was used to compare the RFS between the high and low T cell responders and the ROC analysis was performed using a logistic regression model. SAS v9.4 and R v4.4.3 were used to create Fig. 1 and Extended Data Figs. 2–4 and perform statistical analysis. GraphPad Prism v9.4 was used to create Figs. 1 and 2 and Extended Data Figs. 5 and 6 and perform statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.