
Inclusion and ethics
Table of Contents
The Study was approved by the Institutional Ethics Committee of the Coordinating Center (Sapienza NO. Ref. Ce 5575; Febination 2020) and by the Ethics Committee of EAach Participating Center. The Investigational Sites that approved The Study Protocol Were as Follows: Aou Policlinico Umberto I, Rome; European Institute of Oncology (IEO), IRCCS, Milan; Veneto Institute of Oncology Iov-Irccs, Padua; Division of Medical Oncology, Pisa University Hospital; IRCCS Romagna Institute for the study of tumors (IRST) ‘Dino Amadori’, Meldola; National Institute of IRCCS Foundation ‘G. Pascale ‘, Naples; IRCCS National Cancer Institute Regina Elena (IRe), Rome; IRCCS Institute of Candiolo, Candiolo; Medical Oncology Unit, Arnas Garibaldi Catania; Santa Maria di Terni hospital, Terni; Pederzoli Hospital, Peschiera del Garda; IRCSS Polyclinic Hospital San Martino, Genoa; Galliera hospitals, Genoa; Central Hospital of Belcolle, Viterbo; IRCCS Sacro Cuore Don Calabria Hospital, Negrar of Valpolicella; Santa Maria della Misericordia Hospital, Perugia; New hospital in Prato-Santo Stefano, USL Toscana Centro company, Prato; Cro Aviano, National Cancer Institute, IRCCS, Aviano; Santa Maria delle Croci Hospital, Ausl Romagna, Ravenna; Oncology Unit Asst Pope John XXIII, Bergamo; Order Mauriziano Hospital, Turin; Aou Policlinico S. Andrea, Rome; Federico II University Hospital, Naples; IRCCS Foundation Institute of Cancer of Milan; BIO-Medical Polyclinic University Polyclinic Foundation, Rome; Ao Papardo-Messina; IRCCS Institute tumors ‘John Paul II’, Bari; ‘Mater Domini’ University Polyclinic, Catanzaro; San Leopoldo Mandic oncological Center, Tiber Island Gemelli Isola; Rome; University Hospital, Ferrara; Misericordia Hospital, Grosseto; Ausl Piacenza Guglielmo from Saliceto Hospital, Piacenza; Comprehensive Cancer Center, Ausl-Irccs of Reggio Emilia, Reggio Emilia; Humanitas Gradenigo, Turin; Foundation IRCCS Casa Relief of Suffering, San Giovanni Rotondo; Ramazzini Hospital, Modena Local Health Unit (AUSL), Carpi; AOUP ‘Paolo Giaccone’, Palermo; and aou of the Marche, Ancona.
The competent authority, Agenzia Italiana del Farmaco (AIFA), authorized the trial on 8 July 2020 (AIFA/SC/P/76132). The trial is registered on ClinicalTrials.gov with identifier NCT04591431 and in the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) with number 2018-002190-21. The trial adhered to the principles of the Declaration of Helsinki regarding research involving human subjects. Forty-one centers received ethics approval and participated in the study enrollment. All patients signed the specifically conceived informed consent form (ICF).
Study design, population and inclusion/exclusion criteria
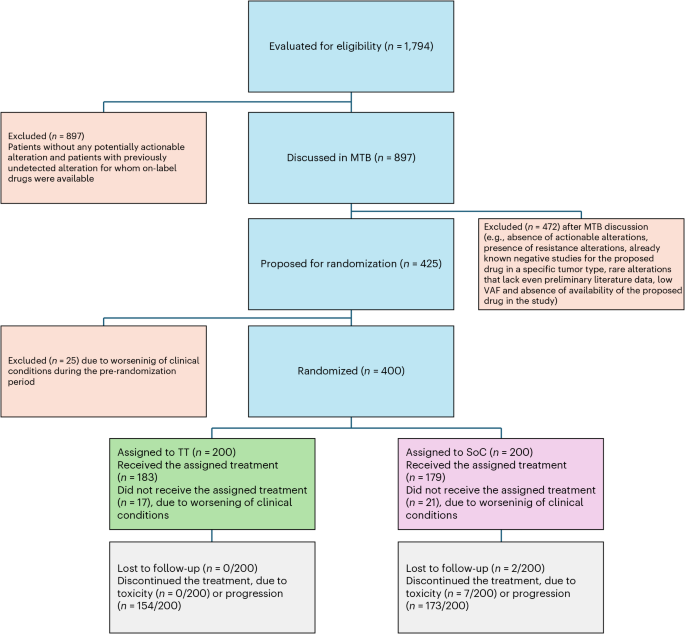
This randomized, prospective, multicenter, proof-of-concept phase 2 clinical trial enrolled patients with inoperable advanced or metastatic solid tumors, regardless of histology. Eligible patients had failed at least one, but no more than two, prior lines of systemic therapy. An ECOG PS of 0 or 1 was required as well as being at least 18 years of age at the time of signing the ICF. All patients had to have measurable or evaluable disease defined per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1)37 or immune-related response criteria (irRC)38. If clinically indicated, magnetic resonance imaging (MRI) and positron emission tomography (PET) scans could have been performed. PET Response Criteria in Solid Tumors (PERCIST)39 were applied in cases where a baseline PET scan was available. Adequate renal, liver and bone marrow functions were required at baseline. Patients who were candidates for potentially curative surgery or other locoregional therapies were excluded. Those with well-established actionable molecular targets, not detected in previous molecular profiling, for which approved and marketed targeted therapies were available—such as EGFR mutations or ALK translocations in lung cancer, BRAF mutations in melanoma, KIT mutations in gastrointestinal stromal tumors, ERBB2 amplification in breast cancer or tumor-agnostic biomarker-driven treatments already available in clinical practice—were also excluded. Furthermore, patients with only bone and/or brain metastases, uncontrolled brain disease (untreated and/or symptomatic) or those whose brain metastases had not been monitored for over 2 months were excluded. Patients with concurrent severe and/or uncontrolled medical conditions that could compromise their participation in the study (for example, uncontrolled diabetes, clinically relevant cardiac disease, uncontrolled hypertension, congestive heart failure, ventricular arrhythmias, active ischemic heart disease, myocardial infarction within the last year, chronic liver or renal disease, active gastrointestinal ulceration or severely impaired lung function) were also excluded (see supplementary materials for all inclusion/exclusion criteria).
For genomic testing, a tissue sample obtained during the screening phase or within 6 months before enrollment was required. The biopsy should be performed during the screening period, after the completion of conventional therapy for recurrent/metastatic cancer. Samples collected within 3 months before the patient’s ICF signature were acceptable, as were samples collected within 6 months, with prior acceptance by the MTB. Archived tissue samples were accepted for patients with glioblastomas and high-grade malignant gliomas. Patients with only one available biopsy, either liquid or solid, due to a failure of one method during the screening phase remained eligible for inclusion and discussion in the MTB. Conversely, if both tissue and liquid biopsy characterizations failed, patients were classified as screening failures.
Molecular profiling was performed using FoundationOne CDX and FoundationOne Liquid CDX tests on tissue and liquid biopsies. DNA from formalin-fixed, paraffin-embedded tumor tissue samples was isolated using the DNA extraction method and analyzed with the FoundationOne CDx panel to assess alterations across 324 genes, including substitutions, insertions and deletions (indels), copy number alterations (CNAs) and gene rearrangements. Genomic signatures of MSI and TMB were also evaluated. For FoundationOne Liquid CDx, circulating cell-free DNA (cfDNA) was extracted from plasma collected from anticoagulated peripheral whole blood samples using FoundationOne Liquid CDx cfDNA blood collection tubes. The cfDNA was analyzed for substitutions, indels in 311 genes, rearrangements in four genes and CNAs in three genes. Additionally, the test evaluated tumor fraction and genomic signatures such as blood-based tumor mutational burden (bTMB) and MSI-H status.
Upon identification of actionable alterations in either tumor tissue or liquid biopsy, and after MTB discussion, selected patients were assigned to receive immunotherapy, targeted therapy or both based on MTB recommendations. A crossover between the two treatment arms was planned upon progressive disease. The study design is reported in Fig. 5.
MTB activities
An MTB convened weekly to review each eligible patient. The MTB consisted of medical oncologists, pathologists, geneticists, immunologists, bioinformaticians and other relevant specialists. Before the MTB discussion, the steering committee identified patients with potentially actionable genomic alterations. The MTB then reviewed each case, considering comprehensive molecular profiling data alongside the patient’s clinical features, such as performance status, comorbidities and concurrent treatments. The treating clinician presented each case selected by the steering committee, highlighting potential actionable therapeutic targets.
Molecular alterations were evaluated using databases such as ClinVAR, OncoKB and COSMIC and the ESMO ESCAT scale. The board employed VAF thresholds of 1% for tissue biopsies and 2% for liquid biopsies. It systematically reviewed potential intrinsic resistance mechanisms, such as KRAS mutations in patients considered for PIK3CA or AKT inhibitors. For patients with multiple actionable alterations (for example, hTMB alongside HER2 amplification or BRAF mutation), the MTB prioritized therapies using a hierarchical framework prioritizing clinical urgency (for example, immunotherapy for MSI-H tumors regardless of co-alterations), followed by biomarker–drug evidence strength (for example, HER2-targeted agents over exploratory TMB-directed therapies when both were present) and, finally, considering toxicity overlap avoidance (for example, excluding PI3K inhibitors if preexisting hyperglycemia).
Patients discussed at the MTB who were not randomized were excluded for various reasons, including the absence of target alterations, the presence of resistance mutations, known negative studies related to a specific alteration, rare mutations without literature data, low VAF, unavailability of drugs for specific alterations, clinical considerations, high levels of uncertainty regarding potentially targetable alterations and the presence of established targetable alterations with available standard treatments. In cases suggesting a germline origin, germline testing was recommended.
MTB recommendations included randomizing patients to receive TT, referring them to a geneticist in the presence of somatic alterations with potential germline significance, modifying SoC only when necessary and suggesting enrollment in clinical trials or early drug access programs, when available in Italy. MTB activities are summarized in Extended Data Fig. 6.
Treatment regimen
Patients who met the inclusion and exclusion criteria and received a recommendation for a specific targeted agent or combination therapy from the MTB were randomized in a 1:1 ratio to receive either SoC or TT, according to their molecular profiles. In the SoC arm, patients were treated with standard chemotherapy, targeted therapy or immunotherapy based on primary tumor histology according to national guidelines and at the investigators’ discretion. The MTB also proposed possible modifications based on molecular profiling and expert opinion in the control group.
In the TT arm, patients received targeted therapy, immunotherapy or a combination that was selected according to their genomic profiles, as determined by the MTB. Combination therapies were proposed in cases where efficacy data for the combination were well established in different malignancies (for example, pertuzumab plus trastuzumab for HER2+ breast cancer and vemurafenib plus cobimetinib for BRAF-mutant melanoma) or when there was a biological rationale due to the presence of multiple actionable mutations. Such proposals were contingent upon the availability of published phase 1b/2 data supporting the combination in other tumor types. In cases involving immunotherapy, the combination of ipilimumab and nivolumab was usually preferred unless a patient’s clinical condition or comorbidities suggested otherwise. The decision was made collaboratively between the MTB and the patient’s treating clinician.
Patients were allowed to crossover to the TT arm upon disease progression. A substantial amendment to the protocol was implemented in October 2022, introducing additional treatments to the existing options, including entrectinib, alpelisib, pralsetinib and selpercatinib. Investigational therapies included erlotinib, pertuzumab, vemurafenib, trastuzumab emtansine, alectinib, vismodegib, cobimetinib, atezolizumab, trastuzumab, ipatasertib, entrectinib, everolimus, palbociclib, lapatinib, ipilimumab, nivolumab, brigatinib, ponatinib, itacitinib, pemigatinib, alpelisib, tepotinib, pralsetinib, talazoparib and selpercatinib. In cases where hTMB coexisted with a targetable mutation, a combination of immunotherapy and targeted therapy was generally preferred, provided that safety data were available. All treatments were administered according to the investigator’s brochure or the relevant regulatory clinical protocol. Each targeted agent was associated with a specific pathway, signature or gene alteration (Extended Data Table 4).
Study endpoints
The primary endpoint of the study was the ORR, defined as the proportion of patients achieving a complete response or partial response, with respect to the total number of randomized patients. Secondary endpoints included PFS, defined as the time from randomization to disease progression or death, whichever occurred first; TTF, defined as the time from randomization until patient withdrawal for any reason, including disease progression or death; TTNT, defined as the time from randomization to the start of the next line of therapy; and OS, defined as the time from randomization to death from any cause. Additional planned secondary endpoints not reported in this paper include concordance between molecular profile on tumor tissue and ctDNA, quality of life measurements, immune fitness in the two treatment arms and the association between the molecular evaluation and gene expression profiling.
Assessment of treatment outcomes and safety
Tumor response was locally assessed according to RECIST version 1.1 and irRC. In patients who discontinued treatment for reasons other than disease progression or consent withdrawal, tumor assessments continued until documented progression. Survival was monitored until death, loss to follow-up, consent withdrawal or study termination. AEs were graded using CTCAE version 5.0, with the frequency and severity of AEs documented for each patient.
Data collection and trial oversight
Data were collected at the clinical sites and inserted into a web-based electronic case report form (eCRF) by investigators or designated personnel. The trial was overseen by an independent external data monitoring committee, which conducted periodic safety evaluations and reviewed efficacy during prespecified interim analyses. Per the study protocol, the data monitoring committee (DMC) conducted a prespecified interim analysis after 20% of the randomized patients (n = 76) had reached the final efficacy evaluation (day 308 or early termination), irrespective of treatment group assignment. The DMC reviewed these data and recommended proceeding with patient accrual as planned. As predefined, the stopping threshold for conditional power (CPt) was set at 5%. Comparisons were made between the observed TT group response rate at interim analysis and the projected end-of-study SoC group response rate (5%). At interim analysis, the TT group exhibited a 7.1% response rate. The calculated CPt for the projected SoC outcome, derived from the sample size assumptions, was 88.7%, demonstrating strong evidence to continue the trial and prompting a unanimous DMC recommendation to proceed. The authors confirm that the trial was conducted in compliance with the protocol, its amendments and the principles of Good Clinical Practice. All authors had access to the data used for manuscript preparation and contributed to its drafting, critical review or editing. The healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945), reviewed this manuscript for medical accuracy only before journal submission. The authors are fully responsible for the content of this paper, and the views and opinions described in the publication reflect solely those of the authors.
Sample size
We hypothesized that TT would yield higher ORR compared to SoC, specifically a 20% ORR for TT versus 5% for SoC. According to the site of primary tumor, four cohorts were defined as breast cancer (Stratum A), non-colorectal gastrointestinal cancers (Stratum B), NSCLC (Stratum C) and ‘other malignancies’ (Stratum D), with competitive enrollment across these strata. To detect a 15% difference between the two arms, assuming an a of 0.10 and a b of 0.20 (80% power) and employing a one-sided chi-square test, a total of 86 patients (43 in each arm of the four cohorts) were required. The four cohorts were evaluated as separate strata, resulting in a combined cohort size of 344 patients (86 patients per stratum). The overall ORR was assessed using a two-sided CMH test at a 5% significance level; an unstratified sensitivity analysis was also performed. Considering a dropout rate of 10%, 384 patients were required. Assuming actionable mutations in at least 30% of eligible patients, the planned screening sample size was 1,280. During the course of the study, the screening failure rate was higher than initially anticipated, leading to a total of 1,794 screened patients. Due to the extended screening periods required for genomic testing and the inherent unpredictability of patient eligibility based on mutational test results, 12 additional patients undergoing screening at the time the target of 384 randomized participants was reached were permitted by the steering committee to enroll, resulting in a final total of 400 patients.
Statistical analysis
All analyses were conducted using SAS software (v.9.4). The SAP, detailing the analyses to be executed, was finalized before database lock (SAP v.1.0). The randomization list was generated with a dedicated SAS program using the PROC PLAN procedure by an independent statistician. The randomization lists were incorporated into the eCRF system, which automatically managed the assignment of treatment arms. A soft lock of the database was performed on 31 July 2024. SAP v.2.0 was finalized on 30 January 2025, after the database freezing and data review meeting, and governs all analyses presented here. The full SAP is available upon reasonable request. Descriptive statistics were used to summarize the data, with mean, s.d., quartiles for continuous variables and frequency distributions for categorical variables. Differences in clinical characteristics were evaluated with t-test, binomial test, chi-quadro and CMH. The Breslow–Day test was used to evaluate the homogeneity of odds ratios. Box plots and tornado plots assessed the overall incidence of grade 3/4 AEs. Kaplan–Meier survival analysis, accompanied by the log-rank test, was employed to compare OS and PFS across arms and subgroups. To visualize clinical benefit duration (response and stable disease) across molecular subgroups—including high TMB and Pi3k/act-altered and ERBB2-positive cohorts—swimmer plots were generated. HRs and 95% CIs were also calculated. Homogeneous subgroups of patients were evaluated in a prespecified exploratory analysis. Exploratory biomarker analyses, including assessments of hTMB (≥10 mutations per megabase), BRAF V600E mutations and HER2 amplifications, were predefined in the SAP to evaluate treatment effect heterogeneity. These subgroups were selected based on emerging preclinical evidence during the trial’s enrollment phase, with steering committee approval for post hoc stratification. All statistical analyses were conducted on the ITT population. R v.4.3.3 and RStudio were used to build toxicity and genomic alterations plots.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.