
Study design and oversight
Table of Contents
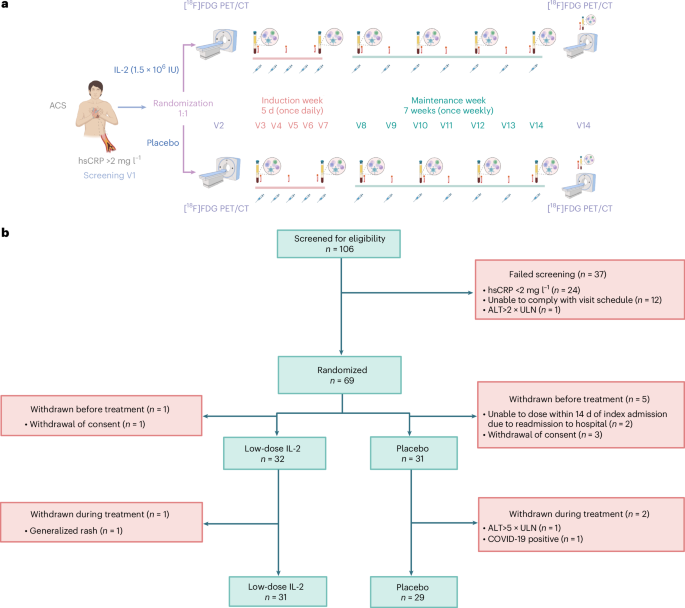
IVORY was a parallel-group, double-blind, randomized, placebo-controlled, phase 2 trial (ClinicalTrials.gov registration: NCT04241601)48. This was an investigator-led trial and the funders had no role in the design, conduct or data analysis of the trial. It was conducted across two sites in the United Kingdom: Cambridge University Hospitals NHS Foundation Trust and Royal Papworth Hospital; 1,383 visits were carried out to complete the trial. The trial protocol was designed by the investigators (see Supplementary Information for the protocol). The trial received favorable ethical opinion from the Yorkshire and Humber—Sheffield ethics committee (19/YH/0171) as well as regulatory approval by the Medicines and Healthcare products Regulatory Authority (MHRA). The trial was conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonisation Guideline for Good Clinical Practice. Written informed consent was obtained from all patients before carrying out any study procedures for both IVORY and IVORY-FINALE. An independent data and safety monitoring committee (M. Marber, M. Dewey, R. Choudhury, G. Lombardi and E. Robinson) reviewed cumulative safety data to safeguard the well-being of the patients.
All patients, clinical investigators and research personnel carrying out study visits or undertaking immune profiling, biomarker and[[18F]FDG PET–CT image analysis were blinded to treatment allocation. Treatment was administered as subcutaneous injections which looked identical (colorless liquid) at equal dose volumes. The PET–CT scans were analyzed with all patient identifiers (including trial identification numbers) and scan dates removed. Therefore, the PET–CT scan analyzers were blinded to the treatment allocation, patient identification details and dates of the scan.
IVORY-FINALE (ClinicalTrials.gov registration: NCT06427694) is a prospective observational study in which cardiovascular clinical outcome data are collected for patients who completed the IVORY trial (Extended Data Fig. 2). The study received favorable ethical opinion from the West Midlands—Edgbaston ethics committee (24/WM/0059) as well as research governance approval by the Health Research Authority (HRA). All prespecified clinical events for patients who completed the IVORY trial were included in this analysis, from the start of the IVORY trial (V1 onwards). The data were collected and analyzed by a team blinded to treatment allocation. A blinded, independent, clinical endpoint adjudication committee reviewed the MACE data.
Public and patient involvement occurred throughout the IVORY trial and this work was also featured on the BBC.
Patients
Adults aged 18–85 years presenting with ACSs were eligible for screening. An ACS was defined as a diagnosis of unstable angina, NSTEMI and STEMI, in patients who had elevated troponin levels or dynamic electrocardiographic changes. A key inclusion criterion was a screening hsCRP level >2 mg l−1.
Major exclusion criteria included presentation with refractory cardiogenic shock or cardiac arrest, those receiving oral or intravenous immunosuppressive therapy, patients with diabetes on insulin therapy and patients with thyroid disorders. Due to the paucity of safety data in pregnancy for low-dose IL-2, women of childbearing potential were excluded. The full inclusion and exclusion criteria can be found in the trial protocol.
Procedures
Patients were randomly allocated to subcutaneously administered placebo (5% dextrose) or 1.5 × 106 IU of IL-2 in a 1:1 ratio using an independent, web-based application (www.sealedenvelope.com). Permutated block randomization was used, with random block sizes of 2, 4 and 6. Randomization was stratified by ST-segment elevation status.
After randomization, patients underwent an[[18F]FDG PET–CT scan of the ascending aorta and carotid arteries in two bed positions, using established and reproducible methods on a dedicated hybrid scanner20,22. The imaging protocol used for the patients can be found in the IVORY trial protocol. Patients were fasted for 6 h before the scans and a dose of approximately 240 MBq of[[18F]FDG was intravenously administered 90 min before image acquisition. The ascending aorta was imaged first, followed by the left and right common carotid arteries. These arterial beds were used because they are the most validated and reproducible in terms of quantifying arterial inflammation in[[18F]FDG interventional drug trials20,21. Arterial[[18F]FDG TBRmax was calculated as the mean arterial maximum standardized uptake value (SUVmax) divided by the average of venous (SUVmean) from both internal jugular veins (for carotids) and the superior vena cava (for the ascending aorta). These methods are detailed further in the IVORY protocol and SAP document.
To ensure accurate serial measures, a number of approaches were employed, such as using a standardized validated imaging protocol with mandated[[18F]FDG uptake times, injected dose and reconstruction parameters, a single dedicated scanner and a phantom study before the start for quantification accuracy.
Dosing commenced within 14 days of admission with ACSs. On completion of dosing, a post-treatment[[18F]FDG PET–CT scan was undertaken. Immunophenotyping (Extended Data Fig. 6) was carried out at the start and end of the induction phase, at alternate visits during the maintenance phase and at approximately 1 week after cessation of treatment. Information with respect to the antibodies used for the FACS can be found in the Reporting Summary. The following antibodies were used according to the manufacturer’s instructions: CD196 phycoerythrin (PE) (20 μl in 100 μl of blood; BD Pharmingen, cat. no. 551773, clone 11A9), CD25 BB515 (5 μl in 100 μl of blood; BD Horizon, cat. no. 564467, clone 2A3), CD194 BB700 (5 μl in 100 μl of blood; BD Pharmingen, cat no. 566475, clone 1G1), CD197 Pe-Cy7 rat (5 μl in 100 μl of blood; BD Pharmingen, cat. no. 557648, clone 3D12), CD185 AF647 rat (5 μl in 100 μl of blood; BD Pharmingen, cat. no. 558113, clone RF8B2), CD4 AF700 (5 μl in 100 μl of blood; BD Pharmingen, cat. no. 557922, clone RPA-T4), CD45RA APC-H7 (5 μl in 100 μl of blood; BD Pharmingen, cat. no. 560674, clone HI100), CD183 BV421 (5 μl in 100 μl of blood; BD Horizon, cat. no. 562558, clone 1C6/CXCR3), CD3 BV510 (5 μl in 100 μl of blood; BD Horizon, cat. no. 563109, clone UCHT1), CD127 BV605 (5 μl in 100 μl of blood; BD Horizon, cat. no. 562662, clone HIL-7R-M21), CD8 BV711 (5 μl in 100 μl of blood; BD Horizon, cat. no. 563677, clone RPA-T8) and CD279 BV786 (5 μl in 100 μl of blood; BD Horizon, cat. no. 563789, clone EH12.1).
Patients who completed the IVORY trial were subsequently invited and then enrolled into the IVORY-FINALE study once written consent had been obtained. A telephone questionnaire was conducted to collect initial clinical outcome data. This was corroborated with medical notes (primary and/or secondary care) before declaration and reviewed by a clinical endpoint adjudication committee.
Outcomes
The primary outcome of the trial was the absolute difference in the mean TBRmax of the index vessel at the end of treatment between the low-dose IL-2 group and the placebo group. This analysis included all segments of the index vessel. The index vessel was defined as the most inflamed vessel (the vessel with the highest mean TBRmax) on the pre-treatment[[18F]FDG PET–CT scans.
The secondary outcomes included the difference in mean TBRmax for more inflamed areas (active segments) of the index vessel at the end of treatment. The threshold for reporting the active segment analysis was predefined as segments with a mean TBRmax > 2 (ref. 49). Additional secondary outcomes included changes in circulating Treg cells, Teff cells and safety or tolerability of treatment and the additive treatment effect of low-dose IL-2 over placebo for various baseline levels of inflammation. The exploratory outcomes included differences between the groups for circulating biomarkers (for example, troponin and hsCRP) and left ventricular ejection fraction.
In the IVORY-FINALE study, the primary outcome was the difference in composite MACEs, including cardiovascular death, nonfatal myocardial infarction, resuscitated cardiac arrest, ischemic stroke or unplanned coronary revascularization, between low-dose IL-2 and placebo at a pre-specified follow-up of 1, 2 and 5 years. The secondary outcomes of the IVORY-FINALE study included differences between the groups in each component of the MACEs, hospitalizations and heart failure, all-cause mortality, amputations and revascularizations for peripheral vascular disease, hemorrhagic stroke, new atrial fibrillation and ventricular arrhythmia.
Statistical analysis
For IVORY, the sample size was based on an absolute difference of 0.2 in the mean TBRmax of the index vessel between placebo and low-dose IL-2 at the end of the treatment period. This is equivalent to a 10% difference from a reference value of 2.02 and similar to the effect observed with high-dose statin therapy32,50. Assuming an s.d. of 0.24, 24 patients per arm, testing at a two-sided 5% significance level, provided 80% power. A sample size of 30 completed patients per arm was selected to account for any uninterpretable imaging or participant dropout. Recruitment ceased once 60 patients had completed the study.
For all continuous variables (including the primary outcome), point estimates and corresponding 95% CIs and P values are reported. For analyses of imaging data, including the primary analysis, linear regression models were used to estimate the treatment effect, adjusting where necessary for baseline covariates and their interactions with treatment. For secondary and exploratory outcomes that were repeatedly measured over time, linear mixed model repeated measures (MMRM) were used to estimate treatment effects for each time point and pooled across time. Where values for each time point are presented, a MMRM analysis with an unstructured covariance matrix for the within-patient residual errors was fitted between V3 and V15. The model specified fixed effects of treatment, baseline value, timing of assessment and an interaction between treatment allocation and timing. For values presented as pooled across time, a second model was fitted that considers only the treatment period up to V15 and does not have an interaction between treatment and time: this estimated the treatment effects averaged over the treatment period. The statistical package used was R. For the MMRM the ‘gls’ function from the ‘nlme’ package was used. The post-hoc analysis for vascular territory–treatment group interactions was done using a generalized least squares model.
For all imaging analyses, ST-elevation status, baseline hsCRP levels and high-dose versus nonhigh-dose statin use were considered for the main effects and interactions with treatment. In any change from baseline analyses presented, baseline values were used as a covariate. Imaging data and T cell data are presented for the full analysis population (as defined in the statistical analysis plan).
To explore the relationship between treatment effect and baseline inflammation, a sensitivity analysis varied the threshold used to define an active segment and 12 threshold values were considered; adjustment for multiplicity was done using the Benjamini–Hochberg process and adjusted and nonadjusted P values are presented for this analysis. A detailed statistical analysis plan can be found in Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.