
DMAb design and delivery platform
Table of Contents
- DMAb design and delivery platform
- Study design and oversight
- Inclusion criteria
- Exclusion criteria
- Safety assessments
- Antibodies
- Quantification of AZD5396 and AZD8076 in serum
- Detection of ADAs against AZD5396 and AZD8076
- Anti-SARS-CoV-2 Spike protein RBD endpoint titer ELISA
- Detection of DMAbs expressed in vivo binding to SARS-CoV-2 RBD
- Pseudovirus neutralization assay
- Purification of DMAbs from participant sera
- Statistical analyses
- Reporting summary
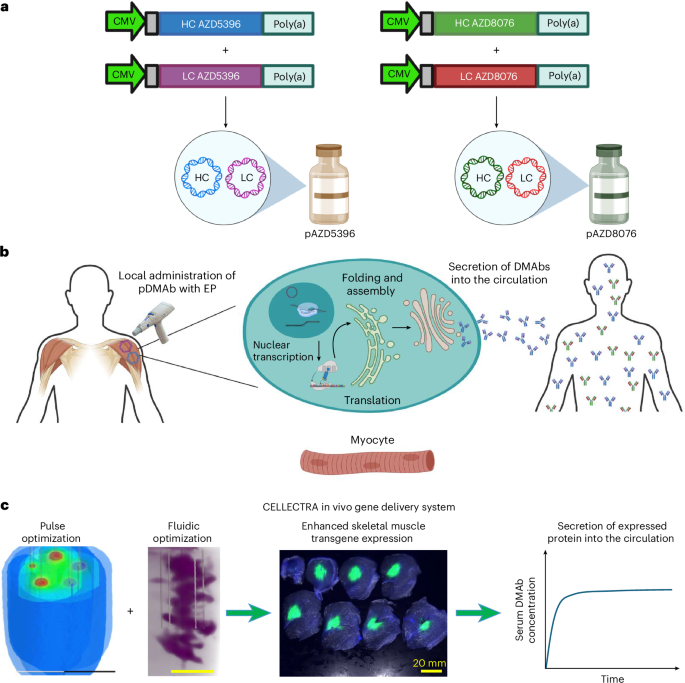
Synthetic DMAb technology was used to enable in vivo production of mAbs targeting SARS-CoV-2. Optimized DMAb constructs were developed in response to the COVID-19 pandemic, encoding AZD5396 and AZD8076, which are based on the parental mAb clones COV2-2130 and COV2-2196, respectively—the precursors of AZD7442. Preclinical studies in mice, hamsters and nonhuman primates demonstrated robust in vivo expression and protective efficacy of this DMAb cocktail12,13.
Each DMAb was encoded by two synthetic plasmids (pAZD5396 and pAZD8076), encoding the HCs and LCs, respectively. The Fc regions of both antibodies were engineered with the YTE (M252Y/S254T/T256E) mutation to extend the in vivo half-life. Plasmids were administered intramuscularly and delivered using the CELLECTRA 2000 with side-port needle in vivo EP technology17,18. The EP system was configured to optimize electric field distribution at the injection site, thereby enhancing plasmid uptake and transgene expression in muscle tissue.
Study design and oversight
This phase 1, open-label, dose-escalation trial was conducted at a single clinical site and approved by the institutional review board (IRB) of the University of Pennsylvania. Enrollment began in May 2022 and was completed in February 2024. All participants provided written informed consent before enrollment.
Eligibility criteria
Inclusion criteria
-
(1)
Age 18–60 years.
-
(2)
Able to provide consent to participate and having signed an informed consent form.
-
(3)
Able and willing to comply with all study procedures.
-
(4)
BMI between 20 and 30, inclusive.
-
(5)
Screening laboratory values within normal limits or with only grade 0–1 findings.
-
(6)
Normal screening ECG or screening ECG with no clinically notable findings.
-
(7)
Women of child-bearing potential who agreed to use medically effective contraception (oral contraception, barrier methods, spermicide) or with a partner who was sterile from enrollment to 6 months after the last injection, or a partner who was medically unable to induce pregnancy. Abstinence was acceptable per investigator discretion if documented and if medically effective contraception was used when engaging in sexual activities, with the study team notified.
-
(8)
Sexually active men considered sexually fertile agreed to use either a barrier method of contraception during the study and for at least 6 months after the last injection, or with a partner permanently sterile or medically unable to become pregnant.
-
(9)
No history of clinically notable immunosuppressive or autoimmune disease. Individuals with HIV infection virologically suppressed for more than 1 year and with current CD4 count greater than 500 cells μl−1 were allowed into the study.
Exclusion criteria
-
1.
Administration of an investigational compound either currently or within 6 months of first dose.
-
2.
Administration of any vaccine within 4 weeks of the first dose.
-
3.
Administration of a SARS-CoV-2 vaccine in the last 90 days or planning to have any standard-of-care vaccines within 14 days from the last administration of the study products.
-
4.
Positive SARS-CoV-2 infection at the screening visit.
-
5.
Administration of any monoclonal or polyclonal antibody product within 4 weeks of the first dose.
-
6.
Administration of any blood product within 3 months of the first dose.
-
7.
Comorbid conditions, including diabetes, hypertension, asthma and any cardiovascular disease.
-
8.
Pregnancy or breastfeeding or planning to become pregnant during the course of the study.
-
9.
Positive serological test for hepatitis B surface antigen or any potentially communicable infectious disease as determined by the Principal Investigator or Medical Director.
-
10.
Positive serological test for hepatitis C (exception: successful treatment with confirmation of sustained virological response).
-
11.
Baseline evidence of kidney disease (creatinine > 1.5 mg dl−1chronic kidney disease stage II or greater).
-
12.
Baseline screening lab with grade 2 or higher abnormality, except for grade 2 creatinine.
-
13.
Chronic liver disease or cirrhosis.
-
14.
Immunosuppressive illness, including hematological malignancy, history of solid organ or bone marrow transplantation.
-
15.
Current or anticipated concomitant immunosuppressive therapy (inhaled, topical skin/eye corticosteroids, low-dose methotrexate or prednisone <10 mg d−1 or equivalent were not exclusionary).
-
16.
Current or anticipated treatment with tumor necrosis factor inhibitors (for example, infliximab, adalimumab, etanercept).
-
17.
Prior major surgery or any radiation therapy within 6 months of the first dose.
-
18.
Any pre-excitation syndromes (for example, Wolff–Parkinson–White syndrome).
-
19.
Presence of a cardiac pacemaker or automatic implantable cardioverter defibrillator.
-
20.
Fewer than two acceptable sites available for intramuscular injection and EP (deltoid and anterolateral quadriceps), including sites with tattoos, keloids, scars within 2 cm of the injection site; implantable cardioverter defibrillator/pacemaker ipsilateral to the deltoid site (unless deemed acceptable by a cardiologist); or metal implants/medical devices at the EP site.
-
21.
Prisoner or participants who are compulsorily detained for treatment of a physical or psychiatric illness.
-
22.
Active drug or alcohol use or dependence that would interfere with adherence to the study requirements or assessment of immunological endpoints.
-
23.
Not willing to allow storage and future use of samples for SARS-CoV-2 virus-related research.
-
24.
Any illness or condition that, in the opinion of the investigator, may affect participant safety or endpoint evaluation.
-
25.
Known bleeding diathesis or use of blood thinners within 30 days before enrollment (low-dose aspirin (81 mg daily) acceptable).
-
26.
Concomitant intramuscular medications.
-
27.
Known previous intolerance or contraindication to methylprednisolone (for participants in cohort D).
Participants were recruited through the University of Pennsylvania Clinical Trials Unit using IRB-approved advertisements, outreach to prior research volunteers and investigator referrals. As enrollment was voluntary, potential self-selection bias is acknowledged; however, this is unlikely to affect the internal validity of the study given its primary objectives of assessing safety and pharmacokinetics in healthy adults.
Study design
Initially designed as a five-cohort dose-escalation study, the protocol underwent multiple amendments in response to U.S. Food and Drug Administration (FDA) and IRB feedback, as well as emerging clinical and procedural data. The protocol was also temporarily amended to allow use of the ProFusion therapeutic needle after concerns about the original needle, which were subsequently resolved. Cohort D was revised to receive a reduced dose without methylprednisolone. As the study progressed, three additional cohorts (E, F and G) were added, expanding the total number to eight. Additional follow-up time points and flexibility in PK/ADA sampling were introduced to improve PK modeling. Based on the tolerability data, EP parameters were changed from OpBlock 0070 to OpBlock 0078 for all cohorts except cohort F.
Participants received intramuscular injections of synthetic DNA plasmids formulated with HYLENEX Recombinant to enhance tissue permeability and improve the efficiency of in vivo transfection, in line with earlier reports17,18. The study followed a single ascending dose design using a modified 3 + 3 schema across eight cohorts (A1–G), with plasmid doses ranging from 0.25 mg to 2 mg and injection volumes from 0.25 to 1 ml. HYLENEX doses ranged from 34 to 135 units, adjusted in proportion to the plasmid dose and volume. Some cohorts received a single injection on day 0, while others followed a more intensive schedule with additional doses on days 3, 28 and 31 (see Fig. 2 and Extended Data Table 1 for cohort-specific details).
Sixty-one healthy adults were screened; of these, 44 participants received at least one dose of the investigational DNA plasmids pAZD5396 or pAZD8076. Sixteen individuals did not meet the eligibility criteria because of timing constraints (n = 5), BMI ineligibility (n = 3), active COVID-19 infection (n = 2), other medical exclusions (n = 4) or personal or external reasons (n = 2). One participant withdrew consent before completing screening (Fig. 2top). Study oversight was provided by an independent Data and Safety Monitoring Board (DSMB).
Sequential enrollment began with cohort A1, where the first participant in each new cohort was dosed and monitored for 14 days before dosing the remaining participants. This waiting period was waived for cohorts D, F and G, where doses were comparable to or lower than those previously tested. Because of the higher dose administered in cohort E, despite lower expression levels observed in earlier cohorts compared to protein-based mAbs, the 14-day observation period was reinstated. In all cohorts, subsequent participants were dosed 3 days apart. If dose-limiting toxicity (DLT) occurred in the first three participants of a cohort, the DSMB conducted an ad hoc review to determine whether to proceed. If deemed unsafe, the remaining participants would have not been enrolled, although the next cohort would not open until 28 days of safety monitoring had been completed. Any dose level with more than one DLT among six participants would have been considered not tolerated. No DLT was observed in the trial.
Each participant underwent a comprehensive baseline evaluation upon consent, including demographic and medical history, concomitant medication review and assessment of eligibility. EP was performed using the CELLECTRA 2000 device. All cohorts were treated using the OpBlock 0078 parameter set, except for cohort F, which used OpBlock 0070. Dosing strategy, plasmid formulation, HYLENEX concentration and the EP settings for each cohort are summarized in Fig. 2.
Safety assessments
Safety was assessed in all participants who received at least one dose of the investigational product (n = 44). Participants were monitored for AEs using the Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials28with laboratory abnormalities evaluated based on site-specific reference ranges. Until the reporting cutoff date, AEs have been documented, including the severity of all related and unrelated AEs (systemic and local) by body system, descriptions of those AEs, elicited local reactions for the first 7 and 10 days after dosing, and the number of individuals who experienced at least one elicited local or systemic reaction in the first 7 days after dosing.
Clinical safety evaluations included vital signs, physical examinations, 12-lead ECG, complete blood counts with differential, serum chemistries, HbA1c, coagulation parameters, serologies, creatine phosphokinase, urinalysis, pregnancy testing (when applicable), PK sampling, ADA assessments and a nasopharyngeal swab for SARS-CoV-2 (Fig. 2b).
The primary safety evaluation focused on the occurrence, nature, timing, duration, intensity and relatedness of both injection site and systemic AEs elicited within 7 days of each administration. Events were categorized using the Medical Dictionary for Regulatory Activities by System Organ Class and Preferred Term. SAEs were collected throughout the study and characterized according to onset, outcome and relatedness to the investigational product. Injection site pain was assessed using a visual analog scale immediately, and at 5 and 10 min after injection/EP, after each dose.
Injection sites (deltoid or quadriceps) varied according to participant preference. Local and systemic elicited AEs were actively monitored for 7 days after each dose. Laboratory safety and PK assessments were conducted at screening, day 0 and day 7, and weeks 2, 3, 4, 5, 6, 8, 12, 16, 24, 52 and 72 after injection. Additional labs were obtained on days 3 and 10 in the two-dose cohorts, and on days 28 and 31 in the four-dose cohorts. All anti-AZD5396 (clone M5B4.2E9) and anti-AZD8076 (clone M16H5.1) AEs, including injection site reactions, were monitored through week 72.
If a predefined safety stopping criterion was met, the study would be paused for review by the DSMB and study team. This did not occur. All safety data were summarized descriptively, with particular attention to potential dose-related trends.
Antibodies
The following antibodies were used in binding ELISAs, pseudovirus neutralization assays and PK/ADA assays.
Commercial antibodies
-
Horseradish peroxidase (HRP)-conjugated goat anti-human IgG (polyclonal) (1:8,000 dilution, cat. no. 2049-05, Southern Biotech).
-
HRP-conjugated streptavidin (1:10,000 dilution, cat. no. N100, Thermo Fisher Scientific).
AstraZeneca research-grade antibodies
-
Anti-YTE monoclonal antibody (clone 23F7.1) used at 1–2 µg ml−1.
-
Anti-idiotype mABs anti-AZD5396 (clone M5B4.2E9) and anti-AZD8076 (clone M16H5), used at 1 µg ml−1.
Anti-YTE mAb (clone 23F7.1), anti-idiotype clone M5B4-2E9 and anti-idiotype clone M16H5.1 were provided by AstraZeneca and have been described previously13. All primary reagents were confirmed using ELISAs. The anti-YTE mAb demonstrated specific binding to YTE-containing AZD5396 and AZD8076 but not to control human IgG serum. Anti-idiotype clone M5B4-2E9 binds specifically to AZD5396; clone M16H5.1 binds specifically to AZD8076. For all ELISAs, recombinantly purified anti-AZD5396 and anti-AZD8076 antibodies were used as standards to confirm reagent specificity.
All antibodies were validated either by the manufacturer, in prior peer-reviewed publications, or in-house during assay development. Full validation details and supporting references are provided in Reporting Summary.
Quantification of AZD5396 and AZD8076 in serum
AZD5396 and AZD8076 were measured using separate ECL bridging immunoassays. For quantitation of AZD5396, 50 µl each of biotinylated anti-idiotypic antibody and SULFO-TAG (ruthenium)-labeled anti-YTE antibody (each at 1 µg ml−1) were mixed in a 96-well polypropylene plate with 50 µl of study serum samples pre-diluted 1:5 in MSD Diluent 100 (Meso Scale Discovery). Plates were incubated on a shaker at 600 rpm for 1 h. In parallel, MSD Streptavidin Gold plates were blocked with 200 µl per well of StartingBlock blocking buffer (Thermo Fisher Scientific) for at least 30 min, then washed three times with PBS-Tween 20. Then, 100 µl of the reaction mix was transferred to the streptavidin plate and incubated for 30 min at 600 rpm. After three washes with PBS-Tween 20, 150 µl of 2× MSD Read Buffer T was added, and the plate was read on an MSD Sector S600 Imager. AZD5396 concentrations were interpolated from a standard curve using recombinant AZD1061 protein (range 1.23–300 ng ml−1 in 20% pooled normal human serum).
The assay for AZD8076 followed the same procedure, except that biotinylated anti-YTE and SULFO-TAG-labeled anti-idiotypic antibodies for AZD8076 were used at 0.25 µg ml−1 each. The standard curve used recombinant AZD8895, which is identical in amino acid sequence to AZD8076 except for the YTE substitutions also present in AZD1061.
Detection of ADAs against AZD5396 and AZD8076
Study serum samples were assessed for the presence of ADAs using a validated three-tiered ECL assay (screening, confirmation and titer), previously validated by PPD for tixagevimab (AZD8895) and cilgavimab (AZD1061), as described in ref. 29. AZD5396 and AZD8076 share identical Fab sequences with their parental mAbs AZD1061 and AZD8895, respectively. Unlike the Fc-silenced versions used in the original AZD7442 product, the DMAbs in this study retained their Fc effector function, bringing the expressed antibodies structurally and functionally closer to native human IgG1. Therefore, the validated ADA assay, which targets the Fab region, is appropriate for detecting immune responses against the expressed DMAbs. The Fc regions also include YTE mutations to extend the half-life, which have not been associated with notable ADA induction in prior clinical studies. For samples with signals below the cutoff points in screening or confirmatory assays, ADA titers were reported as less than 40 for AZD5396 and less than 80 for AZD8076, reflecting the minimum required serum dilution for the respective assays.
Anti-SARS-CoV-2 Spike protein RBD endpoint titer ELISA
Preexisting antibodies against SARS-CoV-2 at study entry (day 0) were evaluated using an ELISA; 96-well plates were coated overnight at 4 °C with 1 µg ml−1 SARS-CoV-2 RBD proteins from ancestral (cat. no. 40592-V08H, Sino Biological), BA.4/5 (cat. no. 40592-V08H130, Sino Biological) and JN.1 (cat. no. 40592-V08H155, Sino Biological) strains. The following day, plates were washed with PBS + 0.05% Tween 20, blocked with Blocker Casein in PBS (cat. no. 37528, Thermo Fisher Scientific) for 90 min at room temperature, then incubated for 1 h with participant sera serially diluted fourfold in blocking buffer. After washing, HRP-conjugated goat anti-human IgG (1:8,000 dilution) was added for 30 min at room temperature. Plates were developed with 1-step Turbo TMB (cat. no. 34022, Thermo Fisher Scientific), stopped with 2N H2SO4 and read at 450/570 nm using a BioTek Synergy Neo2 plate reader (Agilent Technologies). Endpoint titers were determined using the method by Frey et al.30 and analyzed using Excel and Prism v.10.2.1 (GraphPad Software).
Detection of DMAbs expressed in vivo binding to SARS-CoV-2 RBD
Because all participants had preexisting anti-Spike RBD antibodies, a modified capture ELISA was performed to detect DMAbs produced in vivo; 96-well flat-bottom half-area plates (Corning) were coated overnight at 4 °C with 2 µg ml−1 anti-YTE mAb (clone 23F7.1). Plates were washed with PBS-Tween 20 and blocked with 5% nonfat dry milk + 0.2% Tween 20 in PBS for 1 h at room temperature. Sera were diluted 1:4 and serially in twofold steps in 1% newborn calf serum with 0.2% Tween 20 in PBS, and incubated on the coated plates for 2 h at room temperature. Biotinylated SARS-CoV-2 RBD proteins representing wild-type (cat. no. 793906, BioLegend), Delta, BA.2 (cat. no. SPD-C82Eq, ACRO Biosystems) and BA.4/BA.5 (cat. no. SPD-C82EW, ACRO Biosystems) variants were used for detection. After incubation, plates were washed and incubated with HRP-conjugated streptavidin (1:10,000, cat. no. N100, Thermo Fisher Scientific) developed with Ultra TMB (cat. no. 34028, Thermo Fisher Scientific), quenched with 2N H2SO4 and read at 450/570 nm on a BioTek Synergy 2 plate reader. Data were exported to Excel, analyzed using Prism v.10 and reported as AUC over time.
Pseudovirus neutralization assay
HEK 293T cells transformed with SV40 large T antigen (CRL-3216, ATCC) were used for producing the SARS-CoV-2 Spike pseudovirus. Cells were maintained according to the supplier’s recommendations in DMEM supplemented with 10% FCS and 1% penicillin-streptomycin. All experiments involving cell lines were conducted under approved institutional biosafety protocols.
Pseudoviruses bearing the SARS-CoV-2 Spike protein (USA-WA1/2020 isolate) were produced by cotransfecting HEK 293T cells with a 1:1 ratio of pNL4-3.Luc.R-E- plasmid (NIH AIDS Reagent Program) and various Spike-expressing plasmids (GenScript) using GeneJammer (Agilent Technologies). After 48 h, the viral supernatants were collected and supplemented with FCS to a final concentration of 12%.
huCHOAce2 cells (cat. no. VCeL-Wyb019, Creative Biolabs) were plated at 10,000 cells per well in 96-well plates in D10 medium (DMEM + 10% FCS + penicillin-streptomycin), then incubated overnight. The next day, heat-inactivated serum or purified DMAb samples were serially diluted and incubated with pseudovirus for 90 min at room temperature before transfer to huCHOAce2 cells. Plates were incubated for 72 h, then lysed using britelite plus (cat. no. 6066769, Revvity); luminescence was read using a BioTek Synergy 2 plate reader. Neutralization curves were fitted using Prism v.10 (nonlinear regression with Hill slope <0). Minimal infective dose (ID50) values were calculated as the reciprocal dilution yielding 50% neutralization. IC50 values were derived by dividing the serum DMAb titer by the ID50.
Purification of DMAbs from participant sera
DMAbs were purified from serum using anti-YTE-coated Dynabeads (MyOne Tosylactivated, cat. no. 65501, Thermo Fisher Scientific). Day 0 or pooled week 24–52 sera were diluted 1:4 in PBS and incubated with anti-YTE Dynabeads overnight at 37 °C. Bead complexes were washed and eluted using Pierce IgG Elution Buffer (cat. no. 21004, Thermo Fisher Scientific). Two elution fractions were collected. For cohorts A1, A2 and D, samples were concentrated using Amicon 30 kDa MWCO (Merck) filters. DMAb concentration was confirmed using ELISA with anti-YTE capture and secondary anti-idiotype mAbs (M54-2E9 for AZD5396, M16H5.1 for AZD8076). Plates were coated overnight with 1 µg ml−1 anti-YTE or 5 µg ml−1 anti-idiotype mAbs, washed, developed using the same protocol as RBD ELISAs and read on a BioTek Synergy Neo2 reader. Data were analyzed in Gen5 and graphed using Prism v.10.2.1.
Statistical analyses
This phase 1 study was not powered for hypothesis testing. Cohort sizes were determined based on prespecified DLT monitoring parameters, consistent with FDA guidance for first-in-human clinical trials. Participants were enrolled in cohorts of n = 3, 5 or 6 depending on dose level, with a target DLT threshold of 30% used to guide cohort expansion or modification.
In addition, smaller sample sizes in the extension phase of the study (cohorts E–G) were primarily determined by the available resources. The goal was to maximize the scientific information obtained, particularly regarding the impact of multisite administration and EP parameters, within the constraints of limited funding and manufacturing capacity. These exploratory cohorts were designed to generate informative PK and safety data to guide future development, rather than to formally test predefined hypotheses.
Statistical analyses were performed using descriptive methods. AEs were tabulated according to dose level, system organ class and preferred term, with frequencies, percentages and exact 90% Clopper–Pearson CIs calculated. Laboratory values, vital signs and other continuous measures were summarized using the mean, s.d., median and range, while PK endpoints, including detection rates and time to a 50% decline from peak concentration, were estimated with corresponding CIs. Disposition, including enrollment, dose administration, study completion and discontinuations, was also described. Interim analyses were conducted in response to emerging safety events; missing data were managed using participant replacement per investigator discretion. No formal power analysis was performed because the study focused on estimation rather than hypothesis testing. All statistical analyses were performed using STATA v.16 (StataCorp). Prism v.10.0 was used for data analysis and graph generation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.